1.- INTRODUCCIÓN
La microscopía láser confocal es una nueva técnica de observación microscópica que está logrando excelentes resultados en diversas ramas de la ciencia (medicina, biología, materiales, geología, etc.) Su éxito se debe a las indudables ventajas que ofrece frente a la microscopía óptica tradicional (imágenes de mayor nitidez y contraste, mayor resolución vertical y horizontal, etc.) y, sobre todo, a la posibilidad de obtener "secciones ópticas" de la muestra, lo que permite su estudio tridimensional.
Aunque el principio de la microscopía confocal fue patentado hace varios años (Minsk, 1957) y los primeros microscopios basados en esta técnica que demostraron su validez fueron descritos por Petran et al. en 1968, su gran aceptación y espectacular desarrollo no ha tenido lugar hasta hace unos pocos años con el desarrollo del láser y de los ordenadores personales.
La mayor parte de las muestras observadas con microscopía óptica son traslúcidas o, en el caso de ser opacas, su superficie de reflexión no se encuentra perfectamente pulida. En ambos casos la luz interacciona con la muestra a varias profundidades por lo que la imagen que llega al observador presenta áreas borrosas debidas a la luz procedente de zonas fuera del plano de enfoque, lo que produce una degradación en el contraste y resolución de la imagen (figura 1 A).

El principio de la microscopía confocal se basa en eliminar la luz reflejada o fluorescente procedente de los planos fuera de foco (figura 1 B). Para ello se ilumina una pequeña zona de la muestra y se toma el haz luminoso que proviene del plano focal, eliminándose los haces procedentes de los planos inferiores y superiores (Boyde, 1988).

2.- BASES INSTRUMENTALES
Parte de la luz procedente de la fuente de iluminación atraviesa un primer diafragma, es reflejada mediante un espejo dicroico y se enfoca en un punto del espécimen mediante la lente de un objetivo. La señal emitida por el punto iluminado (fluorescencia o luz reflejada) vuelve por el mismo camino óptico, pasa a través del espejo dicroico y es enfocada en un detector, un segundo diafragma o pinhole es colocado delante del detector para eliminar las señales procedentes de la zona fuera de foco (Figura 2).
El principio del funcionamiento del Microscopio Confocal se basa en la existencia de dos diafragmas (pinhole), uno entre la fuente de luz y el objetivo y el otro entre el objetivo y el detector. Ambos pinhole deben de estar perfectamente alineados de forma que el segundo de ellos únicamente deje llegar al detector la luz procedente del plano focal. La utilización de un láser como fuente de luz permite focalizar la iluminación en una región muy pequeña de la muestra y con una gran intensidad.
Dado que sólo se ilumina una pequeña zona de la muestra (punto), para poder visualizarla se necesita un sistema de barrido que permita muestrear todos los puntos y un sistema de formación de la imagen donde se recoja la información de cada uno de estos puntos. El sistema de barrido puede ser de dos tipos: que el haz del láser se desplace por la muestra (beam scanning) o que sea ésta la que se desplace, mientras el haz permanece inmóvil (stage scanning) (Wright, et al, 1993). El primer tipo es el más comúnmente empleado, tiene la ventaja de una mayor velocidad de barrido y por tanto de formación de la imagen, además el espécimen no necesita ser movido durante el muestreo por lo que no necesita ser fijado, lo que lo hace especialmente interesante para el estudio de células en vivo. El campo de barrido coincide con el campo de observación del objetivo permitiendo que la zona de estudio pueda ser localizada utilizando microscopía de fluorescencia convencional
La técnica de desplazamiento de la muestra (stage scanning) presenta comoprincipal ventaja el permitir la observación de una zona tan grande como se desee sin tener que ceñirse al campo visual del objetivo, además debido a que el haz permanece estacionario se tiene una iluminación axial constante.
La luz reflejada o fluorescencia emitida por la muestra es recogida en un fotomultiplicador donde se transforma en una señal de vídeo que se digitaliza y almacena en un ordenador, visualizándose a través de un monitor. La mayoría de los sistemas cuentan con varios fotomultiplicadores y un sistema óptico que permite recoger en cada uno de ellos diferentes longitudes de onda.
Este tipo de microscopio confocal en el que el haz del láser barre la muestra es denominado Confocal Láser Scanning Microscopy (CLSM). Debido a que el láser necesitaun tiempo para barrer la imagen, ésta no pueda ser visualizada de manera instantánea en el monitor.
El método de trabajo del microscopio confocal es por epiluminación, es decir con muestras que al incidir la luz sobre ellas reflejan toda o parte de la luz incidente (microscopía de reflexión), o emiten luz en una longitud de onda superior (microscopía de fluorescencia). El primer caso se suele utilizar con muestras opacas, principalmente en estudios de materiales, mientras que la fluorescencia se utiliza principalmente con muestras biológicas
3.- LA FLUORESCENCIA
Se denomina fluorescencia a la propiedad que tienen ciertas moléculas de, al absorber luz de una determinada longitud de onda, emitir luz en una longitud de onda superior. La fluorescencia puede darse de forma natural en determinadas sustancias (clorofila, algunos tejidos frescos, etc.), denominándose fluorescencia primaria o autofluorescencia. En otros casos para que la muestra que queremos observar tenga fluorescencia es preciso teñirla con un marcador fluorescente, denominado fluorocromo. En este caso hablaríamos de fluorescencia secundaria.
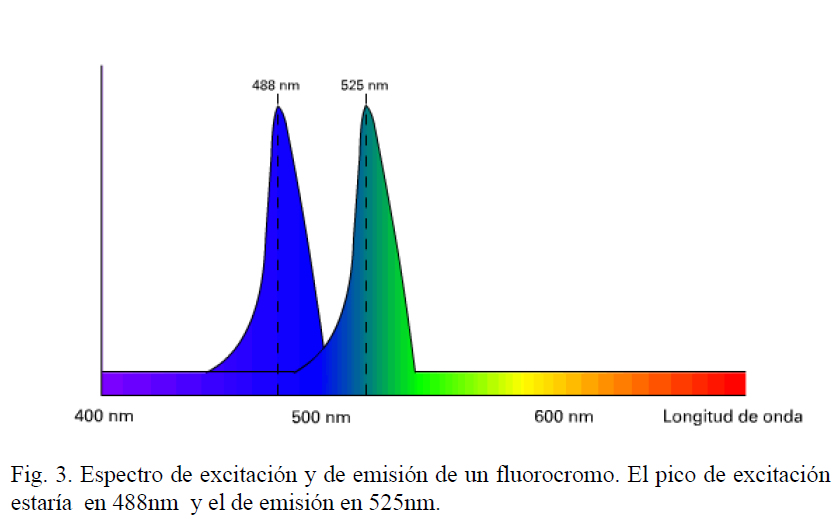
Existen una gran cantidad de fluorocromos que permiten marcar de forma selectiva la mayoría de los componentes celulares y tisulares, además podemos asociarlos a proteínas o anticuerpos para estudiar funciones celulares.Todos los fluorocromos vienen caracterizados por su espectro de excitación y su espectro de emisión (figura 3). El espectro de excitación es el rango de longitudes de onda en las que un fluorocromo absorbe luz, o lo que es el mismo es excitado. El espectro de emisión es el rango de longitudes de onda en las que un fluorocromo emite luz. Ambos espectros presentan dos picos que se corresponden con la máxima absorción y la máxima emisión
En la Figura 4 se muestra el esquema de un microscopio láser confocal Leica TCS SP2 AOBS (Leica Microsistemas). Su funcionamiento es como sigue: De las distintas líneas de láser que tiene el equipo el selector de excitación AOTF nos permite seleccionar la que deseamos utilizar, el haz de láser atraviesa un filtro óptico acústico AOBS y atravesando el objetivo, ilumina un punto de la muestra. Un conjunto de espejos galvanométricos permite desplazar el láser por toda la zona de muestra. La señal luminosa emitida vuelve por el mismo camino óptico, atraviesa el filtro AOBS siguiendo un camino distinto al del haz incidente y llega a un sistema de detección espectral donde es dividida en función de sus longitudes de onda. Un diafragma delante del detector espectral permite seleccionar la luz procedente del plano focal. Esta luz incide en un fotomultiplicador donde es transformada en una señal eléctrica que se digitaliza mediante un convertidor analógico digital y se almacena en un ordenador. La imagen del espécimen es visualizada en la pantalla del ordenador a medida que el láser barre toda la zona de muestra. Una platina con enfoque motorizado permite variar de forma automática la posición del plano focal.

A la hora de adquirir imágenes con el microscopio confocal deberemos de ajustar los
siguientes parámetros:
- Línea de excitación e intensidad del láser. Normalmente un microscopio confocal viene con varios láser que tienen una o varias líneas de emisión: Argon (458, 476, 488, 496 y 514nm), Helio-Neón (543nm), Helio-Neón (633nm), Diodo azul (405 nm), etc. Un filtro óptico acústico (AOTF) permite seleccionar la línea o líneas de emisión que mejor se ajusten al espectro de excitación de los fluorocromos que tenemos en la muestra. Este filtro permite también regular la intensidad del láser. A mayor intensidad del láser tendremos mayor emisión de fluorescencia, pero también se nos producirá un mayor photobleaching (disminución de la fluorescencia con el tiempo).
- Apertura del pinhole (diafragma) de detección. Para cada objetivo existe una apertura óptima del diafragma de detección conocida como Airy 1 en la que el espesor de la sección que se obtiene es el mínimo lo que garantiza la máxima resolución en el eje Z. Cuando la intensidad de la fluorescencia es baja es posible que necesitemos aumentar la apertura de este diafragma para recoger más señal. En este caso estaremos aumentando el espesor de la sección y perdiendo resolución en Z.
- Ganancia del fotomultiplicador. Ajusta la amplificación de la señal eléctrica generada a partir de los fotones emitidos por la muestra. Si la intensidad de luz emitida por la muestra es débil deberemos de aumentar la ganancia para poder obtener la imagen. Un aumento excesivo de la ganancia se traduce en una
pérdida de calidad de la imagen debido al ruido electrónico que se genera.
- Velocidad de barrido del láser. Se define como el número de líneas por segundo que barre el láser. A menor velocidad de barrido mejor relación señal/ruido y por tanto más calidad de imagen.
- Tamaño de la imagen. Define el número de píxeles que tendrá la imagen. Cuanto mayor sea el tamaño de la imagen para un campo determinado mejor será la resolución de la imagen
5.- VENTAJAS DE LA MICROSCOPÍA CONFOCAL.
Las principales ventajas de la microscopía confocal frente a la microscopía óptica tradicional son las siguientes:
- Mayor resolución. Para un objetivo de inmersión en aceite con una apertura numérica de 1.4 y una longitud de onda de 442 nm es posible alcanzar resoluciones de 0.14 μm en horizontal y 0.23 μm en vertical (Wilson, 1990)
- Mayor contraste. Debido a que se elimina la luz procedente de las zonas fuera de foco.
- Posibilidad de realizar secciones ópticas. Variando el plano de enfoque el sistema es capaz de tomar imágenes a diferente profundidad. Lo que permite obtener información tridimensional de la muestra.
- Análisis de imágenes. Al obtenerse la imagen de modo electrónico es posible digitalizarla y aplicar sobre ella toda una serie de técnicas de análisis de imágenes como: realce de imágenes, para mejorar su calidad, combinación de imágenes para comparar cambios en el tiempo, medida de intensidades, medidas morfométricas, etc.
- Reconstrucción 3D. A partir de las secciones ópticas es posible aplicar técnicas de reconstrucción 3D que nos permitan visualizar las estructuras.
- Imágenes multidimensionales. El microscopio confocal nos permite estudiar imágenes en 2 y 3 dimensiones a lo largo del tiempo. Es posible programar el equipo para obtener imágenes durante un periodo de tiempo determinado.
- Imágenes Lambda. Si el equipo cuenta con un detector espectral podremos tomar imágenes a diferentes longitudes de onda (lambda scan) y a partir de ellas deducir el espectro de emisión de un fluorocromo determinado. El sistema de barrido punto a punto (scan) utilizado en los microscopios confocales de tipo CLSM comporta una serie de ventajas adicionales como son:
- Posibilidad de obtener imágenes perpendiculares al plano XY tomando la misma línea a diferentes profundidades.
- Fijar el láser sobre un punto o una pequeña zona de la muestra y tomar imágenes a diferentes tiempos para observar los efectos del láser sobre esa zona.
- Aumentar la resolución mediante zoom del área a barrer tomando mayor número de puntos en áreas más pequeñas
La velocidad de formación de la imagen depende también del número de puntos que tomemos por imagen, así es mayor en imágenes de 512 x 512 píxeles que en imágenes de 1024 x 1024 píxeles. Se denomina pixel (del inglés picture element) a cada uno de los puntos que forman la imagen
En la práctica las grandes aperturas se utilizan cuando se tiene una señal débil de fluorescencia, sacrificándose en este caso la confocalidad por una mayor intensidad de la fluorescencia en la imagen.
La resolución de la imagen es otro de los factores, como se ha visto anteriormente, que afectan a las prestaciones del sistema. Una mayor resolución de la imagen implica una menor velocidad en la formación de la misma, por lo que en aquellos casos en que sea necesaria una rápida adquisición de la imagen (por ejemplo, visualización de procesos dinámicos que cambian rápidamente con el tiempo) será necesario tomar imágenes de menor resolución, pero con un tiempo de formación mucho más rápido.
La resolución de la imagen esta condicionada también por las capacidades de proceso y almacenamiento del sistema informático del microscopio. Una imagen de 1024 x 1024 píxeles ocupa 1 Mbytes de información, esto significa que una serie compuesta de 36 secciones, con esta resolución, ocupará 36 Mbytes de información, necesitándose alta capacidad de almacenamiento en disco y alta velocidad de proceso para poder trabajar con estas imágenes.
7.- PROCESO DE IMÁGENES Y MICROSCOPÍA CONFOCAL
El hecho de que la mayoría de los microscopios láser confocal utilicen un sistema informático para digitalizar las imágenes facilita la aplicación de técnicas de proceso y análisis de imágenes.
La visualización de muestras con fluorescencia débil puede ser mejorada mediante la acumulación de un determinado número de imágenes. Un fondo con ruido debido a una alta amplificación de la señal puede ser corregido mediante promedio de varias imágenes. Otras técnicas, como realce de contornos, filtros, etc., pueden ser aplicadas a las imágenes de microscopía confocal para realzarlas. En estudios de cinética celular los cambios experimentados en un determinado componente (Ca++, por ejemplo) pueden visualizarse mediante sustracción de imágenes tomadas a diferentes tiempos.
Para trabajar con microscopía de fluorescencia, ya sea convencional o confocal, se ha de tener en cuenta que la longitud de onda de la fuente de iluminación (el láser en el caso del confocal) se corresponda con la longitud de onda de excitación del fluorocromo utilizado. La eficiencia es máxima cuando la longitud de onda del láser coincide con el pico del espectro de excitación del fluorocromo (figura 5).

Por lo tanto antes de decidirnos a utilizar un fluorocromo determinado debemos de estar seguros de que alguna de las líneas de láser de nuestro microscopio confocal coincide con su espectro de excitación. La intensidad de la señal fluorescente será mayor cuanto más próxima esté la línea del láser al pico de excitación del fluorocromo.
En la figura 6 puede verse una imagen de unas fibras de pasta de celulosa observadas con microscopía de fluorescencia tradicional y con microscopía láser confocal. Las fibras se observan más claras en la imagen confocal debido a su mayor resolución y a que la fluorescencia de los planos fuera de foco ha sido eliminada.

8.2.- Imágenes teñidas con varios marcadores fluorescentes (multiple labeling).
Múltiples estructuras de una célula o tejido pueden ser observadas simultáneamente tiñendo la muestra con dos o más marcadores fluorescentes, donde cada fluorocromo marca una determinada estructura o sirve como indicador de determinados procesos celulares.

El microscopio confocal suele venir equipado con uno o varios láser que emiten en diferentes longitudes de onda pudiendo utilizarse varias de ellas como fuente de iluminación de la muestra. Si tenemos una muestra con dos o tres marcadores fluorescentes podremos recoger simultáneamente su señal en diferentes fotomultiplicadores.
La figura 7 muestra la configuración del microscopio Leica TCS SP2 AOBS para observar una muestra de bacterias ( Streptomices Antibioticus) marcadas con SYTO13 y Ioduro de Propidio. Se han escogido como fuentes de iluminación las líneas de láser de 488 nm y 543 nm. La señal del primer marcador se recogerá en el fotomultiplicador 1 (PMT1) que se ha programado para recoger un rango de señal entre 490 y 535 nm. El fotomultiplicador 2 (PMT2) recogerá la señal del segundo marcador en un rango entre 580 y 700 nm. Como puede observarse los rangos de detección de ambos fotomultiplicadores se han ajustado para que coincidan con los espectros de emisión de los fluorocromos.
El marcador SYTO13 (verde) tiñe la pared celular de las bacterias mientras están vivas. Cuando mueren este marcador es reemplazado por el Ioduro de Propidio y las bacterias aparecen en color rojo (figura 8) (Manteca et al, 2005).

8.3.- Realización de secciones transversales (x-z).
En la figura 9 se muestra un esquema de las diferentes secciones que podemos estudiar con el confocal (x,y ) ó (x-z) y un ejemplo de secciones longitudinales y transversales de una fibra de pasta de celulosa (Gónzalez-Río et al 1997).

8.4.- Información tridimensional de la muestra.
Como se ha mencionado anteriormente una de las mayores ventajas de la microscopía confocal es la posibilidad de estudiar tridimensionalmente la muestra a partir de secciones ópticas de la misma. Para ello se fija la posición de barrido del microscopio en un extremo de la estructura a medir y se van tomando imágenes, correspondientes a diferentes secciones de la misma, hasta llegar al otro extremo. En la figura 10 puede observarse un conjunto de 30 secciones ópticas de un grano de polen. La distancia entre secciones es de 5 μm.

Integrando las imágenes tomadas en los diferentes planos focales, es posible obtener una imagen de la información tridimensional en foco (Figura 11). Matemáticamente esta imagen, denominada extended-focus, vendría dada por:
I EF (x, y)= ∫ I(x, y,z)dz

La construcción de una segunda imagen extended focus con un pequeño desplazamiento de las secciones permite obtener un par estereoscópico que visualizado adecuadamente proporciona una visión tridimensional de la muestra (figura 12).

8.5.- Reconstrucción tridimensional.
La imagen estereoscópica suministra información tridimensional de la muestra desde un punto de vista estático y en una determinada dirección. Un método mejor para analizar y estudiar las complejas relaciones espaciales es la realización de reconstrucciones tridimensionales donde el volumen de datos puede ser visualizado desde varios ángulos (figura 13).

8.6.- Estudios de actividad intracelular (ph e iones).
Debido al tiempo que se precisa para adquirir la imagen, en aquellos procesos en que los cambios en la actividad iónica son muy rápidos es preciso tomar imágenes de menor resolución o incluso captar únicamente unas pocas líneas por imagen para poder observar la evolución en tiempos muy cortos.
8.7.- Determinación de espectros de fluorescencia (lambda scan).
Si el sistema cuenta con un detector espectral es posible utilizarlo para obtener el espectro real de emisión del fluorocromo con que hemos marcado nuestra muestra. Aunque existen múltiples tablas de los espectros de emisión de los fluorocromos, hay ligeras variaciones entre el espectro teórico y el real debidas principalmente a variaciones en el PH al preparar la muestra o a variaciones de temperatura.
Para obtener el espectro de emisión definiremos un rango de valores de longitudes de onda dentro del cual vamos a tomar imágenes, a continuación definiremos el número de imágenes y finalmente el intervalo para cada imagen. Como resultado tendremos una serie de imágenes que nos muestran la intensidad de fluorescencia para cada intervalo. Si realizamos una gráfica de las intensidades obtendremos el espectro real de emisión de ese fluorocromo (figura 14). Este método es útil también para deducir espectros de autofluorescencia.

9.- NUEVOS DESARROLLOS.
Uno de las principales limitaciones de los microscopios confocales de barrido punto a punto es el tiempo de obtención de las imágenes (entre 2 y 3 imágenes por segundo). Esta lenta velocidad de adquisición supone un problema cuando se trata de observan organismos en movimiento (por ejemplo bacterias) o procesos de dinámica celular que ocurren en un corto intervalo de tiempo. En la actualidad ya existen equipos en los que el láser incide simultáneamente en toda una línea de la imagen en lugar de en un único punto obteniéndose velocidades superiores a 200 imágenes por segundo. Estos microscopios denominados Confocal Live, precisan de equipos informáticos con una gran capacidad de proceso y almacenamiento para poder manejar los cientos de imágenes que podemos obtener en unos pocos segundos.
Wilmer J Sánchez V-19358601
CAF - Parcial 3
No hay comentarios:
Publicar un comentario